Fenilcetonuria
La fenilcetonuria es un trastorno metabólico hereditario que
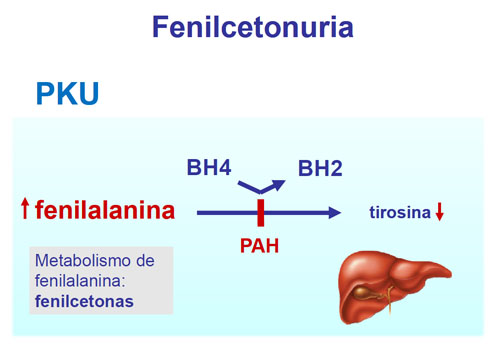
se caracteriza por la carencia o la baja presencia de una enzima, la
fenilalanina hidroxilasa o PAH, que es necesaria para convertir la fenilalanina
en otras sustancias que el organismo necesita. La fenilalanina es un
aminoácido, que son los que forman las proteínas, y por tanto está presente en
la mayoría de alimentos. Los individuos afectados por esta enfermedad no son
capaces de transformar la fenilalanina en tirosina, un aminoácido esencial en
el proceso de formación de los neurotransmisores. La acumulación de
fenilalanina es tóxica para el sistema nervioso, por lo que si esta enfermedad
no se trata a tiempo se pueden producir daños cerebrales y retraso mental.
Los síntomas de la fenilcetonuria varían en severidad. El
tipo de fenilcetonuria más grave se conoce como fenilcetonuria clásica. En este
caso, los niños aparentan estar sanos al nacer, pero si no reciben el
tratamiento adecuado, padecen retrasos en el desarrollo intelectual. También
pueden padecer otros síntomas como convulsiones, problemas de comportamiento o
desórdenes psiquiátricos. Los niños con este trastorno suelen tener la piel
clara y el pelo más claro que el resto de la familia, así como desórdenes en la
piel como eczemas. Otras formas más leves de este trastorno tienen menos riesgo
de daño cerebral.

No hay comentarios:
Publicar un comentario